This web page was produced as an assignment for Genetics 677, an undergraduate course at UW-Madison
Introduction
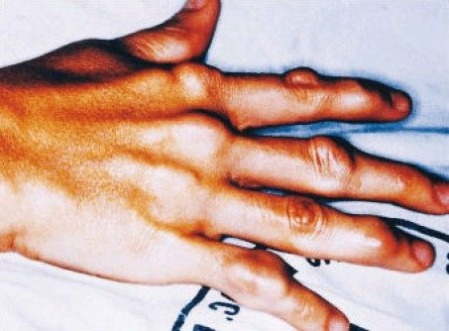
Figure 1-Xanthoma formation as a result of high LDL levels in patient with FH (2).
Familial hypercholesterolemia (FH) is an autosomal dominant disease characterized by extremely high blood cholesterol levels, which leads to premature cardiovascular disease (CD). Individuals with this disease are unable to uptake low-density lipoprotein (LDL), a specific type of cholesterol, from the blood because of certain defective proteins involved in the cholesterol clearance pathway. Sufferers of FH usually die from CD associated myocardial infarction (heart attack) in their 30s or 40s, however severe cases may see these effects in their teenage years. Another symptom of FH is the visible deposition of cholesterol under the skin, known as xanthomas (1).
Low Density Lipoprotein Receptor Gene--LDLR
Patients with FH have mutations in their LDL-receptor (LDLR) gene. Located on chromosome 19 (19p13.2)*, LDLR has 18 exons that encode for the 860 amino acid protein (3, 4). The translated protein has a 21 amino acid signal sequence at the N-terminus that is cleaved once the LDLR protein is transported correctly. Therefore, the functioning protein consists of 839 amino acids (5). The LDL-receptor binds and internalizes LDL from the blood by interacting with apolipoprotein B-100 (apoB), a protein associated with LDL and lipoprotein particles (1).
*Discrepancy between LDLR location: Entrez gene lists it at 19p13.3 while Ensembl and reference 6 have the gene located at 19p13.2. It seems that the general consensus for LDLR location is 19p13.2. Although Entrez gene lists the location at 19p13.3, its MapViewer link shows the gene's location in 19p13.2. 19p13.2 will be used as the default location of LDLR.
*Discrepancy between LDLR location: Entrez gene lists it at 19p13.3 while Ensembl and reference 6 have the gene located at 19p13.2. It seems that the general consensus for LDLR location is 19p13.2. Although Entrez gene lists the location at 19p13.3, its MapViewer link shows the gene's location in 19p13.2. 19p13.2 will be used as the default location of LDLR.
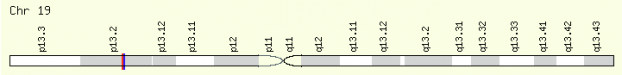
Figure 2-Chromosomal location of LDLR. Click for link to GeneCards website.
Prevalence and Phenotype
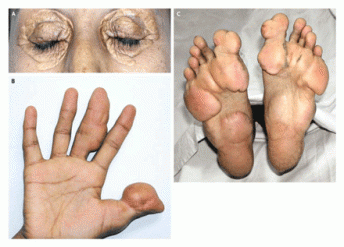
Figure 2-Multiple xanthomas in 40 year old female patient with FH (8).
FH can be caused by at least 900 mutations in LDLR with differing disease severity depending on how crippling the mutation is to the receptor’s function and how many mutated gene copies are inherited (1, 6). A mutation database can be found here. Because FH is a dominantly inherited, one mutated copy of LDLR produces the disease phenotype. This form of the disease is known as heterozygous FH and is treatable with statins or other drugs and a diet low in cholesterol and fat. Individuals with this form of FH usually live into their 40s before succumbing to CD and its complications. If two defective copies of LDLR are inherited, known as homozygous FH, the disease is much more severe. The most accepted treatment for homozygous FH is LDL apheresis, which mechanically filters LDL from the patient’s blood. Individuals with homozygous FH can develop CD by their teens and most die before 30. Heterozygous FH is fairly common, occurring in 1:500 people, while the homozygous form is much more rare, only affecting 1:1,000,000 people (1). Some populations have been shown to have an increased frequency of FH: Afrikaners of South Africa, French Canadians, Finns, and the Christian Lebanese (1, 7). The focus of this website will be on analyzing LDLR using bioinformatics in order to make connections between FH and this receptor.
References
1. Rader, D. J., Cohen, J., & Hobbs, H. H. (2003). Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. The Journal of Clinical Investigation, 111(12), 1795–1803. doi: 10.1172/JCI200318925
2. Photograph of hand xanthomas. Retrieved February 7, 2010 from: http://www.scielo.br/scielo.php?pid=S0066-782X2000000700005&script=sci_arttext
3. National Center for Biotechnology Information. LDLR low density lipoprotein receptor. Retrieved January 28, 2010 from: http://www.ncbi.nlm.nih.gov/gene/3949?ordinalpos=1&itool=EntrezSystem2.PEntrez.Gene.Gene_ResultsPanel.Gene_RVDocSum
4. National Center for Biotechnology Information. Protein-low density lipoprotein receptor precursor. Retrieved February 4, 2010 from: http://www.ncbi.nlm.nih.gov/protein/4504975?report=genpept
5. Yamamoto, T., Davis, C. G., Brown, M. S., Schneider, W. J., Casey, M. L., Goldstein, J. L., Russell, D. W. (1984). The human LDL receptor: a cysteine-rich protein with multiple Alu sequences in it mRNA. Cell, 39(1), 27-38. doi: 10.1016/0092-8674(84)90188-0
6. Leigh, S. E., Foster, A. H., Whittall, R. A., Hubbart, C. S., Humphries, S. E. (2008).Update and analysis of the University College London low density lipoprotein receptor familial hypercholesterolemia database. Annals of Human Genetics, 72(4), 485-98. doi: 10.1111/j.1469-1809.2008.00436.x
7. National Center for Biotechnology Information. Online Mendelian Inheritance in Man-Low Density Lipoprotein Receptor. Retrieved February 2, 2010 from: http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=606945
8. Girish, M. P., & Gupta, M. D. (2005). Images in clinical medicine. Xanthomatous pseudospectacles in familial hypercholesterolemia. The New England Journal of Medicine, 352(23), 2424. PMID: 15944427
2. Photograph of hand xanthomas. Retrieved February 7, 2010 from: http://www.scielo.br/scielo.php?pid=S0066-782X2000000700005&script=sci_arttext
3. National Center for Biotechnology Information. LDLR low density lipoprotein receptor. Retrieved January 28, 2010 from: http://www.ncbi.nlm.nih.gov/gene/3949?ordinalpos=1&itool=EntrezSystem2.PEntrez.Gene.Gene_ResultsPanel.Gene_RVDocSum
4. National Center for Biotechnology Information. Protein-low density lipoprotein receptor precursor. Retrieved February 4, 2010 from: http://www.ncbi.nlm.nih.gov/protein/4504975?report=genpept
5. Yamamoto, T., Davis, C. G., Brown, M. S., Schneider, W. J., Casey, M. L., Goldstein, J. L., Russell, D. W. (1984). The human LDL receptor: a cysteine-rich protein with multiple Alu sequences in it mRNA. Cell, 39(1), 27-38. doi: 10.1016/0092-8674(84)90188-0
6. Leigh, S. E., Foster, A. H., Whittall, R. A., Hubbart, C. S., Humphries, S. E. (2008).
7. National Center for Biotechnology Information. Online Mendelian Inheritance in Man-Low Density Lipoprotein Receptor. Retrieved February 2, 2010 from: http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=606945
8. Girish, M. P., & Gupta, M. D. (2005). Images in clinical medicine. Xanthomatous pseudospectacles in familial hypercholesterolemia. The New England Journal of Medicine, 352(23), 2424. PMID: 15944427
